 ARB is a database program for sequence data, alignments and trees. It is primarily used by the microbial rDNA community, although it is equally powerful for other genes and taxonomic groups. ARB is my primary productivity software for phylogenetics and I thought I would introduce it briefly.
ARB is a database program for sequence data, alignments and trees. It is primarily used by the microbial rDNA community, although it is equally powerful for other genes and taxonomic groups. ARB is my primary productivity software for phylogenetics and I thought I would introduce it briefly.
Although it has some irritations, and took me a little effort to install and learn, ARB is the most powerful phylogenetic environment currently available. Yes, there are some great phylogenetic inference softwares (I like RAxML, PhyML) but that isn’t the same thing at all. This is an environment for understanding sequence data and associated information in a phylogenetic context, not just inferring a good tree. My workflow runs something like this-
- Search GenBank for sequences from taxonomic group of interest.
- Import entire GenBank records into ARB (add my own sequences)
- In ARB, align using Clustal and build quick NJ tree
- Use ARB to add group names (e.g. “Nematoda”) from GenBank/EBI taxonomy or use my own group names (e.g. “very small worms”)
- Check alignment belonging to “weird looking” phylogenetic groups and branch lengths, edit where necessary
- Export alignment as newick file and build a good ML tree in RAxML
- Re-import ML tree to ARB and transfer group names from previous annotated tree
- Ponder
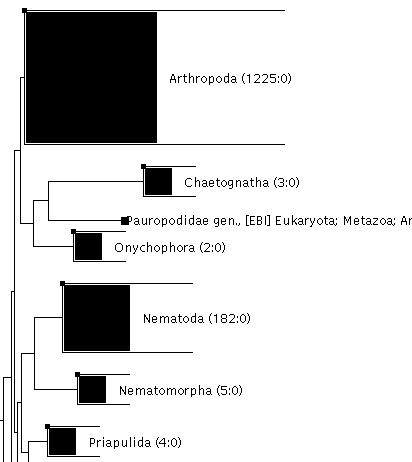
 Can you imagine having 10,000 sequences in an alignment editor? How long would it take you to check the alignment and make minor corrections? What about the tree of those 10,000 sequences? Are the names sensible? Can you include the accession numbers, or contract the genus name to a single letter without rebuilding the tree? How long does it take to scroll through your tree? What taxonomic info are you actually seeing when you scroll, is it just a blur of names, are you just relying on your memory of what species you are looking at or is there node labelling and group collapsing to help?
Can you imagine having 10,000 sequences in an alignment editor? How long would it take you to check the alignment and make minor corrections? What about the tree of those 10,000 sequences? Are the names sensible? Can you include the accession numbers, or contract the genus name to a single letter without rebuilding the tree? How long does it take to scroll through your tree? What taxonomic info are you actually seeing when you scroll, is it just a blur of names, are you just relying on your memory of what species you are looking at or is there node labelling and group collapsing to help?
Although ARB is far from perfect it is powerful and well designed. I don’t really see alternatives out there for dealing with lots of sequences and keeping all the data about those OTUs accesible. Its what I’m going to be using to explore building (and understanding) trees with lots of tips.
I’m actually using a very old version at the moment. There was a new version released in December 07, but I’m waiting for my new machine to arrive before I install it. I’m looking forward to checking it out.
