tl;dr
Phylogenetic experiments need explicitly designed reproducibility, rather than accidental or partial reproducibility. There are many working reproducibility solutions out there differing in their approach, interface and functions. There is no perfect solution for all cases, and you can learn a lot by investigating.
Here I discuss a few software approaches to reproducible phylogenetics. I’m not sure that it is possible for me to review each of these here. Instead consider it a list of bookmarks for you to see the much better examples and descriptions at their home websites. I’ll make some brief comments about their suitability for reproducible phylogenetics though please remember that your experience may differ from mine, and I have only relatively briefly investigated each.
Sumatra
“Sumatra is a tool for managing and tracking projects based on numerical simulation or analysis, with the aim of supporting reproducible research. It can be thought of as an ”automated electronic lab notebook” for simulation/analysis projects”
The extensive Sumatra documentation refers mostly to the command line usage but I really like the GUI that runs out of a web browser tab. Data provenance is managed for you, with the ID of information recorded. Version control is via git or similar though not really incorporated into Sumatra itself, instead Sumatra nags when trying to use data not present or changed in the git repository. I wish this was a little more frictionless, but it might be my lack of experience. A typical design would be to run a script with a parameter file and I’m not sure of its flexibility with more complex arrangements.
make files
Make files are popular in some circles (Broman: “Minimal make”) and can certainly help achieve reproducibility. I can’t help feeling though that this is a direction only for the computationally very experienced. It doesn’t seem likely that this is the way forward if the goal is to increase community reproducibility. What is the best approach is irrelevant unless it is used, and make is unlikely to be broadly used.
phyloGenerator
phyloGenerator (Pearse & Purvis 2013) isn’t primarily reproducibility software, but is a phylogenetics pipeline that describes itself as:
“an easy way for ecologists to make realistic, tenable phylogenies. One-click install and fully-customisable”
I include it here because it’s a great piece of work by Will Pearse and is a wonderful example of a phylogenetic pipeline. Even if many aspects of explicit reproducibility are missing it is considerably closer to reproducible phylogenetics than standard approaches.
ETE-NPR
ETE-NPR is a really smart and powerful package from Jaime Huerta Cepas and the ETE team, describing itself as:
“providing a complete environment for the design and execution of phylogenomic workflows”
It uses the Nested Phylogenetic Reconstruction approach of Huerta-Cepas et al, 2014. One thing I particularly like is the portability of the software as the package makes use of Vagrant to allow standardisation of the environment. Like phyloGenerator, ETE-NPR isn’t reproducibility software, but the workflow design means that it is closer than most approaches and could become so.
Taverna
Taverna (Wolstencroft et al. 2013) is one of the leaders in reproducible workflows, and there is great documentation and videos available. I do not wish in my own work however to use web resources for analysis, they are too unreliable. If you feel differently then Taverna is perhaps for you. One of the things I love most about Taverna is the easy sharing of workflows via the MyExperiment website. Need to build a workflow? Someone will have almost done it before and posted it for all to use, now you only need to tweak a workflow.
Knitr & Sweave
The R community has lead the way in creating reproducible experiments, and sweave and knitr are often involved. Pweave is a python version of sweave. These generally create documentation of the experiment by using the ability to mix markdown and code in a single system. Since the actual code is found in the report the experiment can be reproduced if data is also archived. These seem like mature systems, but I don’t use R much, and I can’t quite see the advantage over IPython notebooks (below).
IPython notebooks
I am very impressed by IPython notebooks for user-friendly reproducible science. The notebooks are a mixture of cells, which can be code cells or documentation cells. Code is not just Python, but also 15 other languages including R, Julia, or even system shell commands. Code runs in an interactive way, with outputs appearing in the notebook instantly under the script that generated them. There are even interactive widgets to modify parameters and see its effect on the data in real time, via sliders. It has a large and active user community, is under active development, and is widely used by scientists of all disciplines. For me the best thing however is that it is the most user friendly of all scripting solutions. The documentation cells mean that you can create something resembling a basic GUI in many ways: “Change example.fas below to your real sequence file name, then press run to get a histogram of the sequences GC content”.
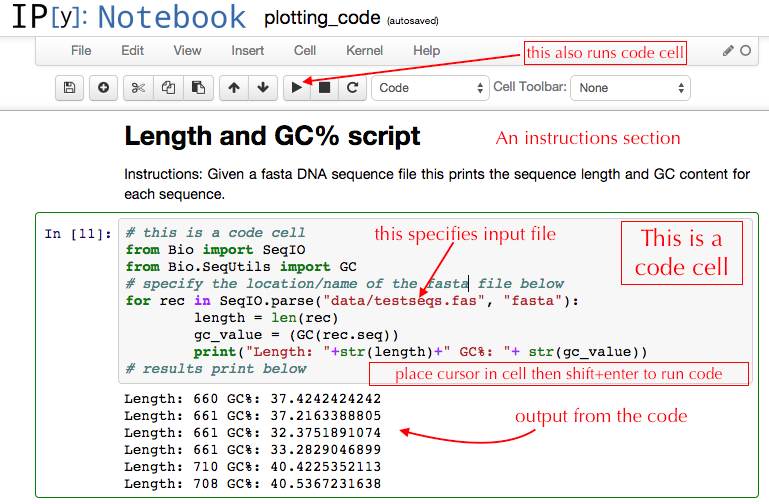
I have really noticed how quickly students and postdocs take to scripting in IPython notebooks. Its interactive nature is a very intuitive way to write and modify code.
Script-based phylogenetics means that all parameters and the instructions for data transformations must be reproducibly recorded. Integrating version control like git can solve the recording of all data file changes, and even explicit provenance is possible.
The future of IPython Notebooks probably lies in Project Jupyter which evolved from IPython, is language agnostic, and is being developed with the Julia community and Google. Google have been developing Colaboratory which I am really excited about. It uses Jupyter and Google drive with all the sharing, commenting, and collaboration options that gives.
This Video introduces Jupyter and then Colaboratory (about 9 mins for both talks).
Galaxy
Galaxy (Goecks et al. 2010) is probably the most powerful and usable solution for reproducibility with a large and vibrant developer community. It is primarily used for genomics but one of the most interesting solutions so far is Osiris (Oakley et al. 2014) from Todd Oakley’s lab. This leverages the built-in reproducibility aspects of Galaxy to achieve reproducible phylogenetics. Provenance, data transformation, software environment, and open archiving are all achieved. This is great work. Galaxy certainly can deliver reproducible phylogenetics, but it is not clear that the work will be easy to reproduce outside of Galaxy. Should we be concerned about that? I still feel that there is work to be done though, alternatives to this approach. The fact that Osiris (and ReproPhylo, our solution) are open source though allows much more rapid progress as we discover what works best.
Next…
Next I’ll introduce ReproPhylo. Amir Szitenberg is a postdoc with Mark Blaxter and I and deserves almost all the credit for delivering this reproducible phylogenetic environment. It works with both Galaxy and IPython notebooks. We’ve done some alpha testing, are working on the documentation, and Amir is adding a few last features. Almost finished, almost.
Broman K. Minimal make: a minimal tutorial on make [http://kbroman.org/minimal_make/]
Goecks J, Nekrutenko A, Taylor J, Galaxy Team: Galaxy: a comprehensive approach for supporting accessible, reproducible, and transparent computational research in the life sciences. Genome Biol 2010, 11:R86.
Huerta-Cepas J, Marcet-Houben M, Gabaldón T: A Nested Phylogenetic Reconstruction Approach Provides Scalable Resolution in the Eukaryotic Tree Of Life. PeerJ PrePrints; 2014.
Oakley T, Alexandrou M, Ngo R, Pankey M, Churchill C, Chen W, Lopker K: Osiris: accessible and reproducible phylogenetic and phylogenomic analyses within the Galaxy workflow management system. BMC Bioinformatics 2014, 15:230.
Pearse WD, Purvis A: phyloGenerator: an automated phylogeny generation tool for ecologists. Methods Ecol Evol 2013, 4:692–698.
Wolstencroft K, Haines R, Fellows D, Williams A, Withers D, Owen S, Soiland-Reyes S, Dunlop I, Nenadic A, Fisher P, Bhagat J, Belhajjame K, Bacall F, Hardisty A, Nieva de la Hidalga A, Balcazar Vargas MP, Sufi S, Goble C: The Taverna workflow suite: designing and executing workflows of Web Services on the desktop, web or in the cloud. Nucleic Acids Res 2013, 41(Web Server issue):W557–61.