Our new phylogenomics environment is called ReproPhylo. It makes experimental reproducibility frictionless, occurring quietly in the background while you work on the science. The environment has a lot of tools to allow exploration of phylogenomics data and to create phylogenomic analysis pipelines. It is distributed in a Docker container simplifying installation and allowing the reproducibility of the experimental computer environment in addition to files. I’ve outlined the background to this in previous ReproPhylo posts.
ReproPhylo is not a phylogenomics pipeline it is a reproducibility environment
Well OK it is a phylogenomics pipeline, and I think a very good one, but that was not the primary objective in creating it. We did not set out to make the world’s most sophisticated pipeline for phylogenomics because we felt that was meaningless without reproducibility. There are a number of phylogenomics pipelines out there, and I look forward to learning more from them how they approach phylogenomics. For me, however the starting point has to be reproducibility. We started with the question ‘what would fully reproducible phylogenomics look like?’ and we got to here.
What is reproducibility? and why replication isn’t enough
What is reproducibility? This is important because it helps us to think about workflows, and what we need to achieve them. Firstly, we want to be able to take an experiment we (or someone else) has previously run and re-run it getting exactly the same outputs. This is replication. Secondly, we want to extend or modify the experiment, adding our new sequences perhaps, or using a different tree building algorithm or parameters. This repeat with modification is reproduction. Reproducibility is also used to describe the field, a general term for all types of ‘this sort of stuff’ which is how I usually use it. Replication isn’t enough. Yes we need to be able to repeat the experiment to check that it really works, but then what? Science is about extending other’s work, building on their discoveries, standing on the shoulders of giants to see farther. ReproPhylo has not been designed to just freeze the experiment for replication.
ReproPhylo promotes reproducibility not just replication
An important component of reproducibility is the ability to repeat the experiment with modification. This is the normal scientific process in phylogenetics and phylogenomics, and elsewhere. ReproPhylo promotes this approach, not just by providing the infrastructure to ensure that the experiment can be repeated, but also by providing the user with extensive help in data exploration, to identify loci and parameters for further investigation.
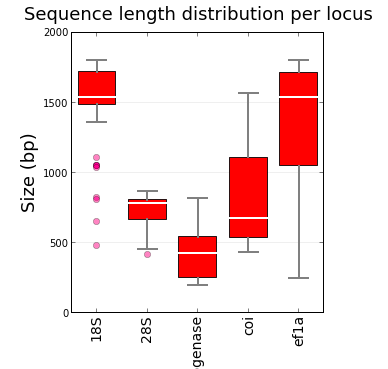
 ReproPhylo helps you to carry out exploratory data analysis (EDA) by providing summary statistics and plots of the raw data and alignments. We have selected a range of these to display, and write an html report characterising the data and results for every analysis without you needing to do anything. Ignore the report, casually browse it, or use it as the basis of your next experiment as you wish. The nature of the stored data, the powerful statistics and plotting modules available in this python environment mean that it is easy to customise your EDA.
ReproPhylo helps you to carry out exploratory data analysis (EDA) by providing summary statistics and plots of the raw data and alignments. We have selected a range of these to display, and write an html report characterising the data and results for every analysis without you needing to do anything. Ignore the report, casually browse it, or use it as the basis of your next experiment as you wish. The nature of the stored data, the powerful statistics and plotting modules available in this python environment mean that it is easy to customise your EDA.
The figure to the left is my version of that by Leek and Peng 2015 discussing p-values. I could have added many more grey boxes. The point is that there is much more to phylogenomics than just how big your support values are, and EDA can help.
EDA is assisted by ReproPhylo presenting many useful plots and summaries. However if you want something else you only have to calculate it once, from then on its available to you, either displayed by default, or when you request it in the pipeline.
Data filtering
When you have a lot of data, a few poor-quality loci may not matter much, might not affect the outcome. But how can you tell? Ideally you should be able to identify loci that are atypical or do not meet some criteria that you specify. Then it should be easy to branch the workflow to exclude loci from the workflow in a separate iteration, and then to test the effect of the change. We do this in the ReproPhylo paper. Does EDA solve all your problems? Probably not, but it gets you to ask questions. In the boxpot do the small outlier loci for 18S make any difference to the analysis? Exclude them and repeat the analysis to test, its easy if the analysis is reproducible and you just need to rerun the script.
Reusability
What is the point of being completely reproducible if it’s just too much work for anyone to actually reproduce your work? I’m sure we’ve all come across situations where we just give up because the goal doesn’t justify the work to get there. I recently spent 2 days of my life trying to open somebody else’s supplementary Nexus format file! Your work should be reusable. This means that it should be easy for someone else to pick up your experiment and extend it, and although reusable is often overlooked it is vitally important. Some of the largest challenges with ReproPhylo were with reusability.
ReproPhylo environments are easy to install
We decided that to be fully reproducible we should try to reproduce the computer environment rather than just the script. This means that when you download somebody else’s workflow you will have the same versions of the software, the same dependencies, and the same ReproPhylo script. It should run identically. This also means that everything is installed in one go.
We have used Docker, the popular container software to do this. I was surprised how easy it was to install Docker ReproPhylo containers. Really it is just one command line to run a shell script. It takes a few minutes the first time (in order to download and install Docker) and is pretty fast thereafter. From that point on you open up the Jupyter (aka IPython) notebook and have everything in place.
Jupyter notebooks make a great interface
I originally thought that ReproPhylo would run primarily in the Galaxy web interface, and we do have it running in this environment. I have been really pleased however with how it looks in a Jupyter notebook. I really like this GUI which is a mixture of explanatory documentation and code snippets (sometimes called literate programming). Our naive guinea pig testers (thanks Dan, Claudia, Max et al.) seemed to take to it well. I wonder if we will maintain the Galaxy interface going forward?
Running ReproPhylo
Is ReproPhylo as easy to use as a binary GUI application like your favourite phylo program? Almost, but not quite, is my honest answer. It is not intended to be, that’s not where we put our effort. It is almost as good though. You can be up and running quickly with one line in the terminal, the instructions for this are written for beginners. Don’t think the documentation is clear enough, or quite right for your OS? Change it! The docs are editable by anyone. Once it’s running open up a notebook that is close to what you want to do from the phylogenetic library, e.g. “Single locus align and ML tree”, or “concatenated alignment with partitions and ML”. (this library is a work in progress, though we already have some written on GitHub) Once you have the notebook open the code snippets to run the analysis are surrounded by documentation explaining, and highlighting the bits to change for your experiment “change this to specify your sequence data”. You should probably just run it to check your data is OK, then tweak the defaults to taste, e.g. swap the aligner, or post alignment trimming or tree parameters. This is much easier than starting with nothing and having to read the damn manual before doing something!
The right balance between naive users, and experienced bioinformaticians and phylogeneticists is challenging. We think that literate programming in Jupyter notebooks is the right approach. Don’t need all the clutter? Delete it and just code your pipeline using ReproPhylo. Lost? Read, ask for help, make explanatory notes for yourself in the notebook, run and repeat. Next time you start with your very detailed notebook and go from there.
What parts of the experiment should be archived and how?
All of it, and in several ways. ReproPhylo achieves reproducibility by a mixture of git, pickle, and saving standard format data files into a Docker container. Amir Szitenberg is going to do a guest post describing some of the underlying workings. At the end of your run, you will have a frozen (pickled) version of the experiment process, that can be easily re-awakened (unpickled) to carry on where you left off. You will have a git repository that has quietly tracked all changes in the background. You will have key outputs (data, alignments, trees) saved in standard text formats (e.g. nexml) that can be imported to other programs outside of ReproPhylo if necessary and a .zip archive of this. The first thing you should do is probably just upload the .zip archive to FigShare, and get a doi. Include the doi in your manuscript, job done, you are now much more reproducible than almost everyone else. To be completely reproducible, and make re-use more likely, you should also archive the Docker container that you have been working in, so storing not just all the data but also scripts and the whole computer environment. It occurs to me writing this that it is idle speculation. The only way to really work out how your experiment should be archived us to have people try, and fail. Archive everything, in several ways, and feed back when things don’t work well.
Computer backups and direct debits
If reproducibility is something you intend to do tomorrow, when you have just finished this analysis, then it has failed. All the evidence suggests that post-hoc reproducibility will be infrequent and unreliable. Computer backups are a good analogy for reproducibility. It is clear that the only strategies that work for backing up your hard drive are ones that do it automatically without requiring your input. All the technical sites agree on this, and however much you really do intend to backup every Friday before you go home, it won’t happen like that. Guilt? That just makes you guilty but no more responsible. Reminders? That works for a short time but then you become desensitised. Automated backup scripts, running in the background, not bothering you but just saving your work to a different drive, yes that works. Businesses know the value of automated action without user input, thats why they are keen on direct debit payments from your bank account, and give rewards for setting them up. They know even good people with the best of intentions are unreliable.
So, can we implement reproducibility quietly, frictionlessly, in the background, without annoying people? Frictionless reproducibility is something I am very keen on. I talk about it a bit in the YouTube video as part of Nick Loman’s Balti&Bioinformatics series. ReproPhylo will take care of reproducibility without you having to do anything except choose to use the ReproPhylo pipeline. You do not need to save your work, or make notes of what file was used, or the settings for that analysis. Just do it, ReproPhylo will record and archive everything, frictionlessly in the background.
If you decide to develop your own reproducibility approach (great!) I very highly recommend that you make it frictionless and don’t put the burden on the user to behave well. I’m really into reproducibility, but I don’t always behave well. We are all just too busy to do this, I want my electricity bill paid automatically, my hard drive backed up regularly and silently, and my phylogenomics reproducibility taken care of for me, without any friction. That way I can concentrate on science.
Here’s something I wasn’t expecting…
ReproPhylo makes your work faster
ReproPhylo is really fast. Not by improving algorithms but by making everything else automated. “Everything else” is the biggest time suck. Example workflows are provided, modest at present, but you can save a library of your own previous analyses, edit the one closest to what you now wish to do and run. You will not have to set up the pipeline from scratch each time. You can also take good ideas from other people’s workflows.
EDA is really important. Most people don’t do it because it’s time consuming. How would you plot a histogram of sequence lengths to check for short ones? How would you identify sequences with a lot of ambiguities? How would you determine if GC content was homogeneous across taxa, or loci? Does your method to do these things scale? ReproPhylo presents you with a lot of information, and it’s not hard to add other things yourself. Want a hexbin plot with marginal distributions of ambiguities vs sequence length? Google it, take a code snippet, and paste it into the notebook. You can’t extend the functionality of standard applications like that!
The future
ReproPhylo is a work in progress, it works and works well, but it does not do everything. It is being developed by Amir Szitenberg, and since we are using it heavily for phylogenomic analyses it is likely to improve steadily. We would love others to use it, and are happy to help. Its all on GitHub as CC0, with public editable documentation. Even if you don’t like our approaches (and we hope you will) you should think very seriously about building your pipeline into a reproducibility environment. You are welcome to make use of ours, you don’t even have to ask. Alternatively contribute to ReproPhylo and make it incorporate your needs.
On our list are many small things, additions that are needed, but the big things are more interesting. ReproPhylo does not yet handle large parallel jobs well and that is going to be needed. Making phylogenomic analysis software scale to increasing data is a major challenge for everyone. Also a challenge is scaling the users’ ability to understand the nature of the big data they are feeding the script for analysis. This is also an important challenge else rubbish in/rubbish out will be a common phrase.
@ReproPhylo is the twitter handle to follow progress.
Webpage and code http://hulluni-bioinformatics.github.io/ReproPhylo/
Documentation is here http://goo.gl/yW6J1J
Preprint http://dx.doi.org/10.1101/019349