My PhD was hard. The grasshopper I was investigating (Chorthippus paralellus) had a huge number of mtDNA insertions into the nuclear chromosomal DNA (numt, pronounced ‘new-might’). PCR tended to amplify multiple templates. It was hard to get genuine mtDNA sequences.
numts are known from almost all animals, though in most species they don’t get in the way of normal PCR ampification. Species that do have a lot of numts, or just a few particularly amplifyable ones, are problematic for mtDNA evolutionary studies.
While working with James Kitson on metabarcoding approaches we started talking about numts, and how they c an be treated in large scale sequencing experiments. James was an excellent postdoc in my lab, shared with Darren Evans, and now a research fellow at Newcastle University. He had come to the project with ideas about using a mixture of bulk Illumina sequencing and amplicon tagging to allow the sequencing of many samples while retaining individual level information (that would not normally be possible with Illumina). We worked on optimising this, and thinking through how it could be used.
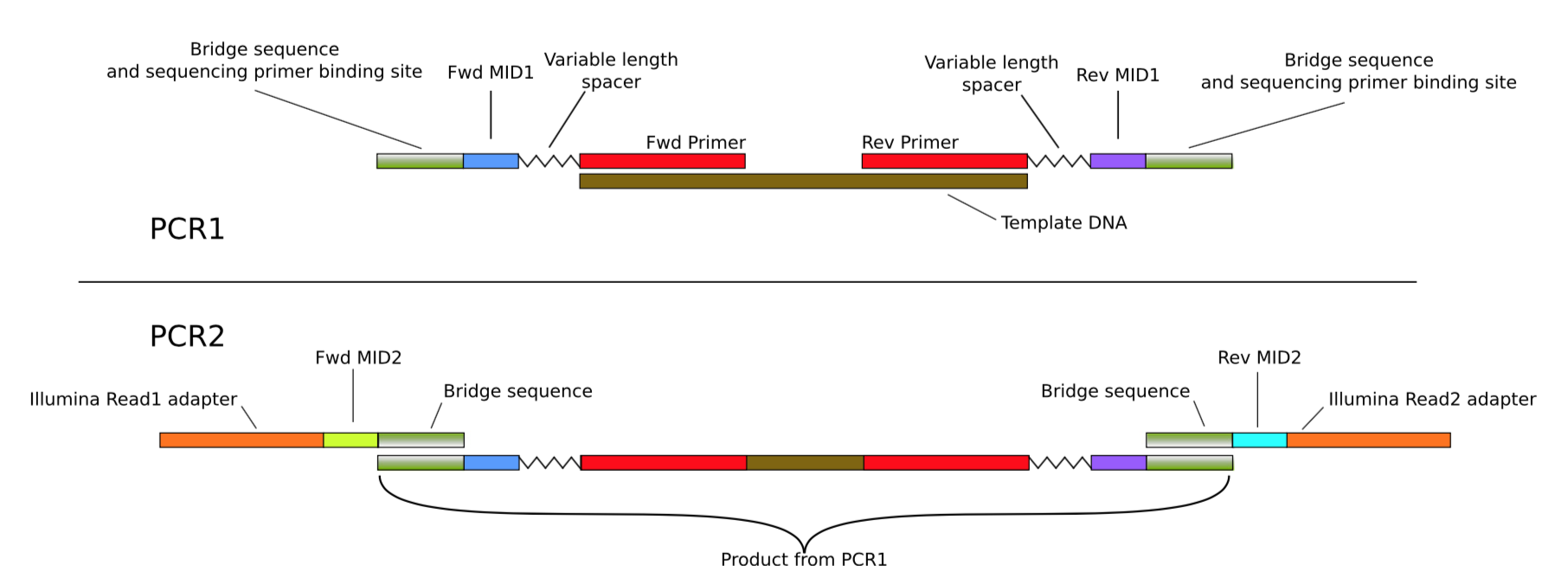
Parasitoid detection by nested metabarcoding
A key part of James’ project was to examine the parasitoids of invasive Oak Processionary Moth caterpillars. Since thousands of reads could be assigned to a single caterpillar he was able to detect both the host and parasite DNA. Through his careful optimisation this approach scaled to a couple of thousand individuals per MiSeq run, and this “Nested Metabaracoding” was written up (Kitson et al 2018). The point wasn’t to say “hey we invented something new” but rather to say that by putting together a number of known approaches a really useful protocol in ecology and evolution could be run in a normal lab. This gave novel ways to look at complex systems, incuding host-parasite systems, and numts.
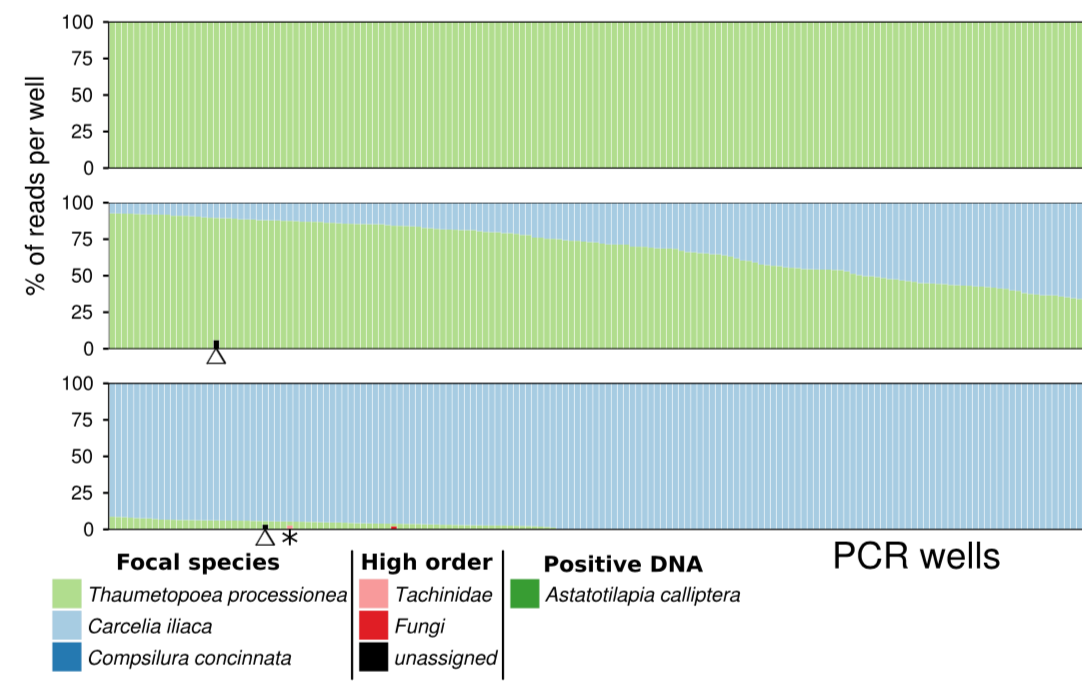
numt detection by nested metabarcoding
Normally, for Sanger sequencing, the ampification of more than one target sequence is very problematic. It leads to messy mixed traces that are always difficult to deal with. Nested metabarcoding however generates many sequences for the same individual but isn’t a problem. The reads can be clustered (or similar) to represent the separate starting templates of host, parasitoid, or numt. It should be relatively straightforward then to exclude numts from further analysis.
How can we know what is numt and what mtDNA? Well evolutionary rate, mutation pattern, and copy number will differ, but ultimately the question is really “which sequences are orthologs?” This approach can separate them and allow proper analysis in this context.
Is this useful?
Yes, very. Whether you think this is useful or not depends on your view of the future of mtDNA analysis. In my opinion there are many projects that benefit greatly from an analysis using mtDNA. Do I think that is all you should do? No. Sequence entire genomes. But to start with you will want to understand the system you are dealing with and mtDNA is a great way to do that. If numt sequences are a problem, nested metabarcoding is a great idea.
Is this cheap?
Yes. If you can get 2000 individuals on a single MiSeq run costs are low. How low I don’t know, it mostly depends on how cheap your tips and tubes and plates are. Plasticware becomes a dominant cost. But its not going to be expensive.
What about nanopores?
Nanopore sequencing is great. Yes it will work also if you can tag your primers appropriately.
Reference
Kitson JJN, Hahn C, Sands RJ, Straw NA, Evans DM, Lunt DH. Detecting host-parasitoid interactions in an invasive Lepidopteran using nested tagging DNA-metabarcoding. PDF Mol Ecol. 2018; doi:10.1111/mec.14518
