Apologies to Jules Verne, but on 30.10.07 there were 20,363 Metazoan SSU rDNA accession numbers in the Silva 92 database. My immediate questions- can a reliable phylogeny be constructed from this by a biologist with average computing resources? Can the resulting tree be informative (or is it too big to get any meaningful information onto a single computer screen)? I think the answer to both is going to be “yes”.
[SILVA92; All domains of life (504,295); Eukarya (73,041)].
The (EMBL) taxonomic breakdown of these Metazoan sequences goes something like this…
48 Acanthocephala; 920 Annelida; 8833 Arthropoda; 27 Bilateria incertae sedis; 57 Brachiopoda; 24 Bryozoa; 30 Chaetognatha; 703 Chordata; 447 Cnidaria; 35 Ctenophora; 21 Cycliophora; 233 Echinodermata; 6 Echiura; 4 Entoprocta; 71 environmental samples; 52 Gastrotricha; 11 Hemichordata; 5 Kinorhyncha; 1 Loricifera; 5 Mesozoa; 2 Micrognathozoa; 1339 Mollusca; 386 Myxozoa; 45 Myzostomida; 4517 Nematoda; 18 Nematomorpha; 64 Nemertea; 7 Onychophora; 8 Placozoa; 1449 Platyhelminthes; 39 Pogonophora; 215 Porifera; 18 Priapulida; 78 Rotifera; 87 Sipuncula; 556 Tardigrada; 2 Xenoturbellida;
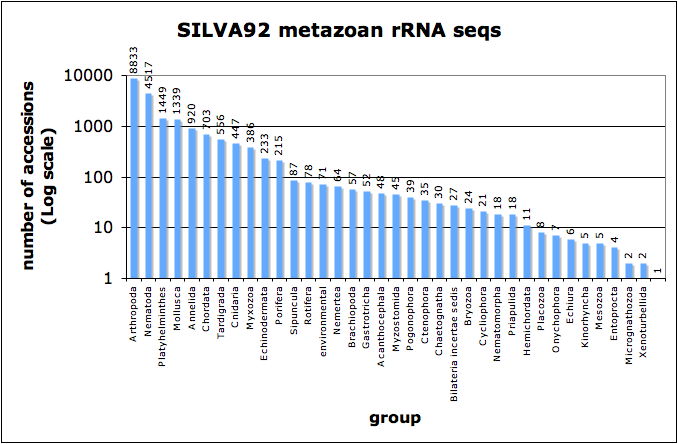 Note that the figure is made with a log scale! There are enormous differences in the sampling effort of different phyla with Arthropoda having almost 9000 sequences and Loricifera just one. These SSU rDNA sequences are aligned using a structural model, and could be an amazing resource for phylogenetics.
Note that the figure is made with a log scale! There are enormous differences in the sampling effort of different phyla with Arthropoda having almost 9000 sequences and Loricifera just one. These SSU rDNA sequences are aligned using a structural model, and could be an amazing resource for phylogenetics.
But there are of course some problems with a dataset of this size, and this is a topic (problems and how to solve or avoid them) I will probably keep returning to.
Why do phylogenetics on 20k sequences?
“Why not just sub-sample a few sequences from each group and do a ‘normal’ tree?”
Well other people have done this before. I think there are a number of issues with this and there are several advantages to “wide and deep” taxonomic sampling in both method and concept. Details will have to wait for another time, but I think it is important and definitely the way people will prefer to go (if it is clear it can be done in a straightforward manner).
This exercise isn’t primarily to redo metazoan taxonomy using a single gene, it is to explore techniques, and determine what is important in taking this approach.
How can we do phylogenetics on 20k sequences?
My primary goal is to be able to use Silva 20k SSU tree for metazoa, construct a decent (and well-annotated) tree, and browse relationships between taxa in an informative way.
I have been playing with tree building approaches on large datasets already, using both minimum evolution and maximum likelihood. More posts on this later. At the moment I have been concentrating on a subset (about 5000 sequences), until I get my thoughts together, and overcome some issues. In summary though, yes it is possible to build trees on this scale on a desktop computer.
What is a “big” tree?
There is no meaningful definition of ‘big’. In the literature when people write “large phylogenetic trees” they have in the past meant 100 taxa, in the recent past maybe 1000 taxa, people now (occasionally) talk about several thousand or more. Obviously the definition changes with time. I just mean a tree too big for the standard software and approaches to deal with comfortably. So that’s more than 1000 (although most software struggles in one way or another with more than about 200 taxa!).
Once you have a tree, how can it be visualised in order to actually learn something?
This is actually a real problem in phylogenetic biology. Viewing very large trees is the topic of an upcoming blog.
I’ve been thinking about constructing, processing and visualising (or extracting info from) big trees quite a bit and have developed some strategies that are very useful to me, and might be to other people too. (I have been routinely dealing with phylogenies of 3-5000 sequences (about 1500bp alignments) on a fairly low spec computer. This post is really to introduce the dataset and the SILVA site and my vague aims.
